Day :
- Pediatric Rare Diseases| Therapies for Rare Diseases| Other Rare Diseases| Current Research on Rare Diseases| Rare Diseases of Immune System| Genetic Diseases and Disorders
Location: London, UK

Chair
Nadia Ameen
Yale University School of Medicine
USA

Co-Chair
Wei Zheng
National Institutes of Health
USA
Session Introduction
Nadia Ameen
Yale University School of Medicine
USA
Title: Ion Transport Defects in Microvillus Inclusion

Biography:
Nadia Ameen completed her medical degree from the University of the West Indies, Jamaica. She completed training in Pediatrics at Children’s Hospital Wisconsin and then pursued post-doctoral fellowship in gastroenterology at Yale University School of Medicine. She is the principal investigator of an NIH supported research laboratory at Yale University that studies CFTR and diarrheal diseases for over 20 years. She was the first to recognize a trafficking defect in MVID before a genetic defect was identified. She has authored more than 30 papers in reputed journals and serves as an editorial board member for several journals.
Abstract:
MVID is a rare congenital disease that results in severe secretory diarrhea (SD) and death in newborns. Brush border (BB) defects, villus atrophy and microvillus inclusions (MVIs) in enterocytes are associated with the diarrrhea. Loss of function mutations in the actin motor Myosin Vb (Myo5b) is responsible for most cases of MVID. How loss of Myo5b results in secretory diarrhea is unknown. The study used Myo5b loss of function human MVID intestine, polarized intestinal cell models of secretory crypt (T84) and villus resembling (C2BBe) enterocytes lacking Myo5b in conjunction with immunofluorescence confocal gSTED imaging, immunohistochemical staining, TEM, shRNA silencing, immunoblots, and electrophysiologic approaches to examine the distribution, expression and function of the major BB ion transporters (Na+ (NHE3), Cl- (CFTR) and Cl- /HCO3- (SLC26A3, DRA) that control intestinal fluid transport. NHE3 and DRA localization and function were markedly reduced on the BBM of human MVID enterocytes and Myo5bKD C2BBe cells, while CFTR localization was preserved. Forskolin-stimulated CFTR ion transport in Myo5bKD T84 cells resembled that of control. Conclusions: Preservation of functional CFTR in immature enterocytes, reduced functional expression of NHE3 and DRA contribute to Cl- and Na+ stool loss in MVID diarrhea.
Wei Zheng
National Center for Advancing Translational Sciences
National Institutes of Health
USA
Title: Drug repurposing screen for rapid identification of therapeutics for Zika virus infection

Biography:
Dr. Wei Zheng received his Ph.D. in Pharmacology from the State University of New York at Buffalo. He had worked 12 years in pharmaceutical companies (Berlex, Amgen, and Merck) for drug discovery and development. Since 2005, Dr. Zheng has worked at National Institutes of Health for development of therapeutics for rare and neglected diseases. He has identified several drugs from drug repurposing screens and advanced two of them to clinical trials. Currently, Dr. Zheng is working on the therapeutics development for lysosomal storage diseases, drug resistant cancer and infectious diseases.
Abstract:
Zika virus infection with complications of microcephaly and other complications has emerged as a health threat in many countries the infection has been reported over 60 countries. Vaccines and therapeutics are currently unavailable for the Zika virus infection. We have developed several compound screening assays and carried out the drug repurposing screens to identify potential therapeutics for the Zika virus infection. The newest results on drug repurposing screens and Zika virus drug development will be presented in this talk.
Diego-Abelardo Alvarez-Hernandez
Faculty of Health Sciences
Anahuac University North Campus
Mexico
Title: Neglected Tropical Diseases: Road to Control, Elimination and Eradication by 2020

Biography:
Diego Alvarez has his expertise on awareness, education, research and fundraising for Neglected Tropical Diseases. As a medical surgeon with interest and passion for infectious diseases, he is currently studying a Master’s degree of Medical Sciences at Anahuac University North Campus while he does laboratory research on Chagas disease at the Department of Microbiology and Parasitology of the National Autonomous University of Mexico. Also he has been working for several years on Antimicrobial Resistance at the Health Sciences Faculty of the Anahuac University North Campus. Nowadays he is working at the Coordination of Medical Services of the Mexican Red Cross PAR Huixquilucan Office and is a former member of the END7 Campus Leaders Council and of the Young Researchers Track of the Global Network for Neglected Tropical Diseases, an initiative of the Sabin Vaccine Institute. He just joined the American Society of Tropical Medicine and Hygiene.
Abstract:
Neglected tropical diseases (NTDs) are a diverse group of bacterial, parasitic and viral diseases that proliferate in tropical and subtropical enviroments through 149 countries. Currently, more than 1.4 billion people living in Africa, America and Asia are affected by at least 1 of the 17 NTDs recognized by the World Health Organization (WHO). NTDs are called “neglected” because they have been largely wiped out of the most developed areas, but they have persisted in the poorest and most marginalized societies, where inadequate sanitation due to the lack of clean water and poor hygiene, frequent contact with vectors and reservoirs and inadequate healthcare services prevail. If left untreated, NTDs may cause substantial illness and tremendous physical and emotional suffering, hampering children from attending to school and reducing adults economic productivity. As a result, families and communities become trapped in a cycle of disease and poverty. Fortunately, NTDs can be effectively managed if proper measures are implemented and while they have been around for centuries, the team effort to fight them is brand new. In 2011, the WHO Strategic and Technical Advisory Group for Neglected Tropical Diseases drew a roadmap for control, elimination and eradication for the 2012-2020 period and in 2012, a community of partners endorsed the London Declaration on Neglected Tropical Diseases to commit themselves to enchance a better and accelerated response by working in alliance. As we consider both events as an historic set point to change the course of NTDs and as we find ourselves at half of the way of the planned period, it is time to analize where does we stand on the roadmap and what areas of improvement should be reinforced. We still can achieve targets, but additional commitment is needed and each health professional should play its role to reach those left behind.
Martine Zimmermann
SVP Global Regulatory Affairs
Alexion Pharmaceuticals Inc
Switzerland
Title: Development of Orphan Medical Products

Biography:
Martine Zimmermann is a Executive Director in SVP Global Regulatory affairs in Alexion Pharmaceuticals Inc located in Switzerland
Abstract:
Overview:
This track will discusses opportunities and challenges associated with developing drugs for rare and ultra-rare diseases, and the use of novel approaches to bridge these challenges and successfully bring important lifesaving therapies to patients in need.
Context/Details:
Between 5,000 and 8,000 distinct rare diseases exist, affecting between around 27 million and 36 million people in the EU. Collectively these diseases represent a significant proportion of the population, and a growing healthcare concern since many of these rare disorders are serious conditions with no approved treatments.
Since roughly 80% of these diseases have identified genetic origin, our scientific understanding of and ability to target these diseases is growing. Technological advances and our increased understanding of underlying disease biology has aided in the development of several groundbreaking therapies over the past ten years. In addition, heightened public policy support and push to find treatments for patients with rare and devastating diseases has helped in the adoption of legislative vehicles and regulatory programs that compliment scientific discovery and provide important development and financial incentives to companies to bring new targets from bench to bedside. Among them include expedited development and approval programs that offer enhanced regulatory dialogue and support for developers from early stages of clinical development through to approval.
More and more we see appreciable regulatory support for innovative program design and increased regulator openness in applying regulatory flexibilities. All these factors have aided in the approval of important medicines for ultra-rare and devastating diseases. Yet challenges do still persist in the development process, chief among them is the absence of knowledge about the disease itself needed to inform important aspects of clinical development. Natural history for rare diseases is often poorly described or missing altogether given the small affected populations who themselves have variable phenotypes and clinical courses. For developers, it is important, early in the development process, to assess the depth and quality of this information so that a parallel study might better inform the development process.
In the this track we will discuss the pathway to development in rare diseases, starting with this most critical first step of assessing and acquiring knowledge of the disease itself through natural history studies. We will discuss endpoint selection, innovative design and leveraging expedited programs to help with small data sets. We will discuss ways in which frequent and early communication is essential. Finally, we will bring all these points together by using a real life example/case study.
Stefano Giacomini
Medical Director
Rizzoli Orthopaedic Institute
Italy
Title: Treatment of Early Onset Scoliosis Associated with Rare Disease

Biography:
Dr. Stefano Giacomini is a Medical Director; performs orthopedic examinations in Free Job regime at the Outpatient Clinic of Rizzoli. He has done his Specialization in Orthopedics and Traumatology achieved in 2001 at the University of Bologna. He is the author of over 50 scientific publications. He has participated, even as a speaker at dozens of national and international conferences and 'socio GIS (Italian Scoliosis Group)
Abstract:
Background
Treatment of non idiopatic spine deformities in young children is very demanding.
Rare syndromes are various clinical conditions, heterogeneous in terms of clinics but associated with spinal deformities in a large percentage of cases. Scoliosis is the most common early onset deformity, sometimes present at birth and often rapidly evolving. Due to the complexity of its clinical aspects and early onset a multidisciplinary and timely approach is mandatory.
The purpose of this study is to describe an approach to spinal deformities in rare syndromes and assess the effectiveness of surgical treatment with growing systems (Growing rod, Magnetic rod and VEPTR-like system) at a young age.
Methods
A retrospective study of 11 paediatric patients (7 females and 4 males) treated at the authors’ department for early onset scoliosis associated with rare syndromes was performed. The patients had been surgically treated for scoliosis with growing systems, i.e. Growing Rod System, VEPTR and GSP, between 2006 and 2010 at the authors’ spine surgery department. Mean follow-up was 24 months (range, 12 to 36 months). Mean age at surgery was 7 years and the patients presented with the following clinical conditions: Escobar syndrome (1), scoliosis associated with congenital heart disease (1), Arnold-Chiari type 1 malformation (1), scoliosis associated with syringomyelia (1), type 1 neurofibromatosis (2), Prader-Willi syndrome (1), trisomy 8 (1), arthrogryposis (2) and spondylo-rib dysplasia (1). Each patient was investigated from the genetic point of view performing the following exams: brain and spine MRI, PFR, cardio-US and abdominal US, neuropsychiatric and neurological evaluation and a specific C0-C1-C2 CT scan to assess any instability, above all when some syndromes, such as arthrogryposis, were involved.
Results
After a total of 11 lengthening procedures, performed 11 months after surgery on average, a correction of the thoracic curve of 63% was observed at the last follow-up and the Cobb angle on average had decreased from 61° to 31°. A total of 8 postoperative complications were encountered, 2 of which required a revision surgery not originally planned.
Conclusions
The growing systems are good devices which have proved effective in the treatment of early onset scoliosis associated with rare syndromes, when deformities are very aggressive on account of the peculiar features of the syndromes and, subsequently, are difficult to be controlled conservatively.
Both clinical and instrumental evaluation performed specifically for each patient were of the utmost importance, since the peculiar features of each syndrome had to be assessed together with the common characteristics of scoliosis shared by all patients.
PFR evaluates the young patients’ life quality indirectly; cardio-US and abdominal US are useful in the study of visceral malformations; brain and spine MRI can detect spinal cord malformation, that is to say contraindications to surgery, and is mandatory for thorough preoperative planning.
The indication for traditional Growing Rod, or Magnetic rod, rather than VEPTR/GSP should be given according to patient’s age and considering the presence of thoracic hyper kyphosis or chest deformities.
Ilham Abuljadayel
Co-Founder
TriStem Corp Ltd
UK
Title: Retrodifferentiation in the Treatment of a Rare Condition: Acquired Aplastic Anaemia

Biography:
Dr. Ilham Saleh Abuljadayel: discovered the process of retrodifferentiation in the early nineties. This direct reprogramming of differentiated somatic cells is achieved through cell surface receptor contact of more mature adult human cells such as leucocytes. She was awarded worldwide patents on the methodology and device, enabling the production of unprecedented levels of pluripotent stem cells from differentiated cells. Based on her research, Dr. Abuljadayel co-founded the TriStem Group. During the period 1990 to 1995, Dr Abuljadayel worked as a consultant immunologist at the King Fahd Armed Forces Hospital in Jeddah, and from 1996-2000 headed the TriStem Research on retrodifferentiation at the London Hospital, Kings College, Downing College University of Cambridge and Addenbrooke Hospital
Abstract:
stem cells can offer cures to treat many rare diseases, which can be used to correct a plethora of genetic conditions or replenish damaged tissue and cells in acquired disorders, in allogeneic or autologous manners, respectively. The limiting factors for such applications are; the availability and quantity of the stem cell source, the identification of a suitable histocompatible donor and the aggressive nature of ablation therapies that enable engraftment . On the other hand, retrodifferentiation technology which is similar to epimorphic regeneration, albeit, occurs ex vivo, offers a rapid additional source of stem cells with high efficiency. The process involves dedifferentiation / retrodifferentiation of mature adult cells such as peripheral leukocytes into a heterogeneous population of stem cells belonging to a give tissue. Retrodifferentiation procedure produces unlimited supply of stem cells from patient or donor blood which have been shown to be safe as well as capable of long term engraftment.
Furthermore, the autologous retrodifferentiated stem cells have been shown to engraft human bone marrow in the absence of ablation, in a rare disease such as acquired aplastic anaemia. This presentation will focus on the production of multipotent stem cells prepared from mononuclear cells and its application in the treatment of aplastic anaemia, a rare condition if left untreated lead to rapid morbidity.
Jorge-Alberto Ascencio-Aragón
Faculty of Health Sciences
Anahuac University North Campus
Mexico.
Title: Current Challenges on Chagas Disease

Biography:
Jorge Ascencio is a medical surgeon with special interest on infectious diseases. He has been doing laboratory and clinical research on antimicrobial resistance and human gut microbiota for several years and recently he started doing laboratory research on Chagas disease at the Department of Microbiology and Parasitology of the National Autonomous University of Mexico. Currently he is working at the Coordination of Health Sciences at Anahuac University South Campus while he studies a Master’s degree of Directorate of Health Services at Anahuac University North Campus. He is a former member of the END7 Mexico Chapter of the END7 Campaign and of the Anahuac University Neglected Tropical Diseases Interest Group
Abstract:
Chagas disease or American trypanosomiasis is recognized by the World Health Organization as one of the 18 Neglected Tropical Diseases. It is estimated that there are between 7–8 million people infected, between 65–100 million people at risk of becoming infected and it causes nearly 12,000 deaths per year worldwide. Chagas disease is endemic of 21 Latin American countries, but the expanding migrational flows have male the disease an international health priority. It is a parasitic zoonosis caused by Trypanosoma cruzi, a protozoan with high genetic and phenotypic diversity that can be principally transmitted to human beings by the faeces of blood-sucking triatomines. Other mechanisms of transmission include; blood transfusions, organ or bone marrow transplants, from mother to child, by ingestion of food or drinks contaminated with triatomine faeces and due to occupational exposure. Chagas disease has a very broad-spectrum of clinical manifestations, depending upon the phase at where the patient is. Acute phase is characterized for passing unnoticed in 95% of the cases, unless Romaña’s sign or chagoma develops at the inoculation site. Chronic phase is characterized for developing cardiac or gastrointestinal disease that lead to increased morbi-mortality. Diagnosis can be done with the combination of epidemiological background and clinical manifestations, if present, but laboratory tests are required for confirmation. Benznidazole and Nifurtimox are the only drugs available for treating the disease and meanwhile the efforts to formulate vaccines remain insufficient, patients suffer from a preventable disease which main risk factor for acquiring it is living in poor and marginalised societies. Control of vector-borne transmission remains to be a challenge in endemic countries as it is related to low socioeconomic status, while serological screening at blood banks and monitoring of all pregnant women for non vector-borne transmission can be effective to control the disease in non-endemic countries
Patrick .J. Tighe
University of Nottingham
UK
Title: Pathway-Centric analysis of rare autoinflammatory diseases (AID) and drug repurposing

Biography:
Dr Paddy Tighe is Associate Professor of Immunology within the School of Life Sciences, The University of Nottingham. Dr Tighes laboratory studies dysregulated immune function and biomarkers associated with a range of chronic diseases with immune system involvement focusing on the orphan autoinflammatory syndromes, and TNF-receptor-associated periodic syndrome (TRAPS) in particular. We were first to report the ligand-independent signaling processes triggered by intracellular accumulation of mutant TNFR1 in TRAPS and our MRC –funded development of high-throughput, high-content methods for intracellular signalling analysis has provided insight into the underlying mechanisms and potential targets for novel therapeutic agents for TRAPS. Current interests are comprehensive mapping of the TRAPS-associated inflammatory phenotype for improved diagnostics and identification of novel therapeutics and associated research into drug-repurposing for TRAPS treatment.
Abstract:
The rare hereditary autoinflammatory periodic syndromes are the result of specific mutations in a range of genes ultimately affecting innate immune response mechanisms, yielding a pro inflammatory state. Despite single genes being causative, the inflammatory consequences are widespread. To better understand the underlying mechanisms associated with TNF receptor-associated Periodic syndrome (TRAPS), as a prototypic model of an AID, we have undertaken extensive examinations of intracellular signalling pathways and cytokines associated with inflammatory responses in such patients, and then applied this roadmap of a perturbed, generally overactive signalling network to seek existing pharmacological drugs which can suppress one or more of the affected pathways, with the goal of repurposing existing drugs for the benefit of TRAPS patients. Several classes of small molecule compound have been identified which show potential benefits, and further classes of small compounds have been derived from in-silico structure and functional comparisons to existing chemical library data.
Yingjun Xie
The Third Affiliated Hospital of Guangzhou Medical University
China
Title: Consulting for a combination of molecular defects for variable expression

Biography:
Xie Yingjun, Ph.D. Working in the Key Laboratory for Major Obstetric Diseases of Guangdong Province,China. Researching in clinical genetic disorders (such as down syndrome, microdeltion/microduplication syndrome, autism, DD and ID).
Abstract:
Background: Expressivity is variable for most of the molecular defects. However, achondroplasia is a well-defined and common bone dysplasia an incidence of approximately 5-15 per 100,000 live births. Gain-off unction mutations in FGFR3 have been shown to cause both chondrodysplasias and craniosynostoses and to result in impaired endochondral ossification
Case presentation
A 2-year-old boy with clinical features consistent with achondroplasia and Silver-Russell syndrome-like symptoms. The patient exhibited features such as scoliosis and a trident configuration of the hands all of which can be explained by a mutations in FGFR3 at c.1138 G > A(p.Gly380Arg). However, prenatal onset growth delay, the speech delay, hypotonia and small triangular face phenotypes were not commonly reported in previous cases of ACH. We further detected a three-fold increase in GRB10 expression. Combining with previous other studies, the one unique feature of this patient that can be directly linked to a GRB10 duplication is the prenatal onset growth delay.
Discussion
The data related to the patient described in the present study at least suggest that mutations in FGFR3 cause ACH but do not influence the effects of the duplication of GRB10 on prenatal onset growth delay in SRS. The results of our study also suggest that phenotypes are rarely “simple” or directly related to specific gene defects and that combinations of uncommon, rare and exceptional molecular defects, which can be explored and used in diagnoses, may explain the so-called variability observed in the expression of dominant traits.
Larissa Kerecuk
Rare Disease Lead
Birmingham Children’s Hospital
London
Title: Implementation of UK Strategy for Rare Diseases: Birmingham Women’s & Children’s Hospital NHS Foundation Trust Engagement with patients & families affected by Rare Diseases

Biography:
Dr Larissa Kerecuk is the Rare Disease Lead at Birmingham Children’s Hospital and is developing the first Paediatric Rare Disease Centre in the UK for holistic patient care. Larissa also leads the 100,000 Genome Project at Birmingham Children’s Hospital.As Consultant Paediatric Nephrologist, Larissa specialises in treating children with kidney diseases including those on dialysis, which require a holistic approach.
Abstract:
At BWCH, everything we do is aimed at improving the care delivered to our patients. Given that we treat approximately 9000 patients with over 500 different rare diseases, it was very important to us to develop a rare disease strategy to address the keys improvement areas highlighted by RDUK. Therefore, BCH are developing the first holistic Paediatric Rare Disease Centre where multidisciplinary and multispecialty rare disease clinics will take place with coordination of care, peer support and better access to research, information and treatment. We are, and have been, consulting with patients and patient support organisations throughout the process. In the initial design process, families have told us that a large waiting area where they can interact with each other and patient support organisations is important so we have incorporated this into the design. We have also included a kitchen for our families to improve the quality of their stay as well to provide a facility for them to feed their children any special feeds that they are often on. Several of our patients have multiple sensory deficits, learning difficulties and/or may have autistic spectrum features so we have also ensured that we have a sensory room and a “chillout room”. As many of our patients and their families are wheelchair bound, all our doors are wider and the toilets are suitable for all our patients – including an adult changing room.
Whilst the rare disease centre is being built, we have applied for funding from the Roald Dahl Marvellous Children’s Charity to fund 3 posts to help to coordinate the care of children with rare diseases. We held a very successful Rare Disease Engagement Events which was very well attended by patients and families with a wide range of rare diseases. The aim of the days was to co-design the posts so that we address the concerns and needs of our families in a way that we can improve their quality of life. We now have 2 Roald Dahl Specialist Rare Disease Nurses and and one Roald Dahl Rare Disease Transition Sister.
Making a diagnosis is very important which is why we are also involved in the 100 000 Genome Project and we are actively recruiting families to this. We aer working with SWAN UK and have developed a bespoke clinic at BCH Rare Disease Centre for children with syndromes without a name.
At BCH, we believe that the first holistic paediatric rare disease centre will help to re-think the care we deliver to patients from a one-size fits all to a patient-centred approach in which we help patients and families with rare diseases lead the best quality of life possible thereby enabling to live their lives to their full potential. We envisage that the rare disease service will lead to development of patient guidelines to empower local teams. Transition to the UHB Rare disease centre will also make the service seamless.
We are also making research an integral part of the care of the children affected with rare diseases and we are able to recruit and discuss research at the same clinic appointment.
The World’s First Children’s Rare Disease Centre will open in December 2017 at Birmingham Children’s Hospital with a new vision in integrated healthcare delivery.
Diego-Abelardo Alvarez-Hernandez
Faculty of Health Sciences
Anahuac University North Campus
Mexico
Title: Neglected Tropical Diseases: Road to Control, Elimination and Eradication by 2020

Biography:
Diego Alvarez has his expertise on awareness, education, research and fundraising for Neglected Tropical Diseases. As a medical surgeon with interest and passion for infectious diseases, he is currently studying a Master’s degree of Medical Sciences at Anahuac University North Campus while he does laboratory research on Chagas disease at the Department of Microbiology and Parasitology of the National Autonomous University of Mexico. Also he has been working for several years on Antimicrobial Resistance at the Health Sciences Faculty of the Anahuac University North Campus. Nowadays he is working at the Coordination of Medical Services of the Mexican Red Cross PAR Huixquilucan Office and is a former member of the END7 Campus Leaders Council and of the Young Researchers Track of the Global Network for Neglected Tropical Diseases, an initiative of the Sabin Vaccine Institute. He just joined the American Society of Tropical Medicine and Hygiene.
Abstract:
Neglected tropical diseases (NTDs) are a diverse group of bacterial, parasitic and viral diseases that proliferate in tropical and subtropical enviroments through 149 countries. Currently, more than 1.4 billion people living in Africa, America and Asia are affected by at least 1 of the 17 NTDs recognized by the World Health Organization (WHO). NTDs are called “neglected” because they have been largely wiped out of the most developed areas, but they have persisted in the poorest and most marginalized societies, where inadequate sanitation due to the lack of clean water and poor hygiene, frequent contact with vectors and reservoirs and inadequate healthcare services prevail. If left untreated, NTDs may cause substantial illness and tremendous physical and emotional suffering, hampering children from attending to school and reducing adults economic productivity. As a result, families and communities become trapped in a cycle of disease and poverty. Fortunately, NTDs can be effectively managed if proper measures are implemented and while they have been around for centuries, the team effort to fight them is brand new. In 2011, the WHO Strategic and Technical Advisory Group for Neglected Tropical Diseases drew a roadmap for control, elimination and eradication for the 2012-2020 period and in 2012, a community of partners endorsed the London Declaration on Neglected Tropical Diseases to commit themselves to enchance a better and accelerated response by working in alliance. As we consider both events as an historic set point to change the course of NTDs and as we find ourselves at half of the way of the planned period, it is time to analize where does we stand on the roadmap and what areas of improvement should be reinforced. We still can achieve targets, but additional commitment is needed and each health professional should play its role to reach those left behind.
- Rare Hereditary Diseases| Rare Ophthalmological Diseases | Genetic Diseases and Disorders |Strategies for Diagnosis &Treatment| Undiagnosed Rare Diseases
Location: London, UK

Chair
Patrick J Tighe
University of Nottingham
UK
Session Introduction
Jong Wook Chang
Samsung Medical Center
South Korea
Title: Paracrine Action of Human Mesenchymal Stem Cells for Muscle diseases.

Biography:
Jong Wook Chang has his expertise in translational and clinical research of stem cells for neurological diseases including CNS and PNS. Especially, he has made effort to identify soluble factors secreted from human mesenchymal stem cells (MSC) to understand therapeutic effect of MSC. In addition, now he is responsible for management of cGMP facility in Samsung Medical Center to produce clinical grade of MSC for clinical trials.
Abstract:
The role of Wharton’s jelly-derived human mesenchymal stem cells (WJ-MSCs) in inhibiting muscle cell death has been elucidated in the present study. Apoptosis induced by serum-deprivation in mouse myoblast cell lines (C2C12) was significantly reduced when the cell lines where co-cultured with WJ-MSCs in a transwell system. Antibody arrays indicated high levels of chemokine (C motif) ligand (XCL1) secretion by co-cultured WJ-MSCs and XCL1 protein treatment resulted in complete inhibition of apoptosis in serum-starved C2C12 cells. Apoptosis of C2C12 cells and loss of differentiated C2C12 myotubes induced by lovastatin, another muscle cell death inducer, was also inhibited by XCL1 treatment. However, XCL1 treatment did not inhibit apoptosis of cell lines other than C2C12. When XCL1-siRNA pretreated WJ-MSCs were co-cultured with serum-starved C2C12 cells, apoptosis was not inhibited, thus confirming that XCL1 is a key factor in preventing C2C12 cell apoptosis. We demonstrated the therapeutic effect of XCL1 on the zebrafish myopathy model, generated by knock down of a causative gene ADSSL1 encoding a muscle isozyme of adenylosuccinate synthase. The exogenous expression of XCL1 resulted in significant recovery of the zebrafish skeletal muscle defects. These results suggest that human WJ-MSCs and XCL1 protein may act as pro-mixing and novel therapeutic agents for treatment of myopathies and other skeletal muscle diseases.
Rob W.J. Collin
Dept. of Human Genetics
Radboud University Medical Center
Title: Splice modulation therapy for inherited retinal diseases

Biography:
Rob W.J. Collin is an Associate Professor at the Department of Human Genetics, at the Radboud University Medical Center in Nijmegen, The Netherlands, and is affiliated to the Donders Institute for Brain, Cognition and Behaviour. During his post-doc, he was involved in the identification of several novel genes underlying inherited hearing impairment, and inherited retinal diseases. Since 2010, he switched his research focus towards the development of Molecular Therapies for Inherited Eye Disorders, in particular those that involve the modulation of pre-mRNA splicing. In 2010, he worked in the lab of Prof. Dr. Jean Bennett, a pioneer in the development of retinal gene therapy. Rob Collin was awarded with a prestigious VENI Award from the Dutch Organization of Scientific Research (2010), an Individual Investigator Award from the Foundation Fighting Blindness (2012), and recently received the IJMS Young Investigator Award (2017).
Abstract:
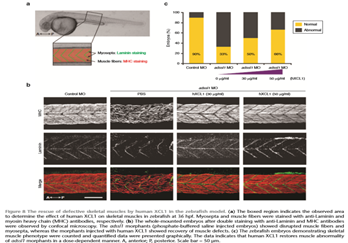
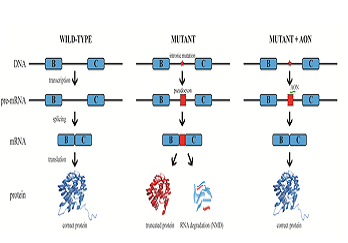
Statement of the Problem: Inherited retinal diseases (IRDs) are characterized by severe and progressive visual impairment, often leading to complete blindness. IRDs display a tremendous genetic and clinical heterogeneity, and to date, no effective treatment exists. A significant fraction of the genetic defects underlying IRD affect pre-mRNA splicing of the corresponding gene. We therefore decided to employ antisense oligonucleotides (AONs) to develop splice modulation therapy for specific genetic subtypes of IRD. AONs are small and versatile DNA/RNA molecules that bind complementary to their target pre-mRNA molecule and are able to redirect pre-mRNA splicing.
Methodology & Theoretical Orientation: Fibroblast cell lines derived from patients harboring mutations that affect pre-mRNA splicing were generated and cultured in the presence of AONs that are specifically designed to correct pre-mRNA splicing. In some cases, fibroblasts were first reprogrammed to induced pluripotent stem cells that were subsequently differentiated to photoreceptor precursor cells, and then treated with AONs. RT-PCR analysis was performed to study the efficacy of AON treatment, and for some cell lines, Western blot analysis or immunocytochemical analysis was performed to determine the effects at the protein and cellular level.
Findings: AON administration to patient-derived fibroblast cells harboring a recurrent splice mutation underlying severe early-onset IRD, i.e. c.2991+1655A>G in CEP290, resulted in full restoration of CEP290 pre-mRNA splicing, a significant increase in CEP290 protein levels, and rescue of a ciliary defect. In addition, AON delivery to patient cells with splice mutations in ABCA4 resulted in correction of the splice defects. Studies targeting additional mutations affecting pre-mRNA splicing that underlie IRD are currently ongoing.
Conclusion & Significance: Splice modulation therapies represent a promising and attractive strategy for the future treatment of specific genetic subtypes of IRD.
Gayathri Balasubramanian
Focus Scientific Research Center (FSRC)
phamax
India
Title: Management of rare diseases: An integrated approach to break down barriers and facilitate patient access to healthcare

Biography:
Dr. Gayathri Balasubramanian is a Scientific Advisor at FSRC, part of phamax AG. She holds a Masters in Clinical Research from the University of Sheffield, UK. She was awarded the “Graduate Award” and “Skills for Work” by the University of Sheffield and other top global organizations for academic, volunteering and extracurricular activities during her tenure there. She has 4 years of progressive experience in the healthcare research domain and she is proficient in primary and secondary research (various assignments employing mixed methods research), systematic reviews and critical appraisal, stakeholders identification, liaison and engagement and publications. She has worked on diverse projects in market assessment, market access, policy shaping, forecasting and epidemiology in therapeutic areas of rare diseases, oncology, diabetes, cardiovascular diseases and infectious diseases.
Email: gayathri.balasubramanian@fs-researchcenter.com
Abstract:
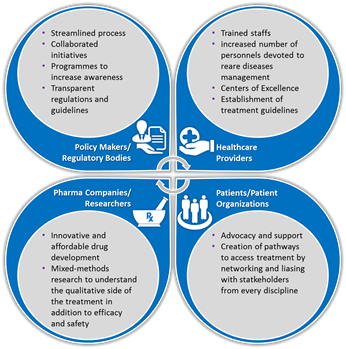
Background: In Europe, a rare disease is a disease that affects less than 1 in 2000. Although this seems to affect a fraction of the population, collectively it imposes a colossal. Rare diseases are disabling, dramatically reducing the quality of life, hampering routine activities and autonomous survival. Rare diseases management is intricate and varies across geographies. Understanding the complexities is important to resolve issues and improve care. Methodology: Secondary research of published and grey literature. Findings: Some of the challenges encountered in rare diseases management are delayed or misdiagnosis, lack of awareness on diseases/management, lack of quality healthcare at proximity, struggles to continued care, treatment access, and social consequences. In recent times, stakeholders such us healthcare providers, patient organizations, pharmaceutical companies, regulatory bodies and policy makers have taken many initiatives to resolve these challenges. National polices/plans are devised exclusively to manage rare diseases. Compassionate use programmes have encouraged research and provided accelerated entry of orphan drugs into the market. Organizations like EURORDIS have given voice to more than 300 rare disease organizations in over 30 countries. While these stakeholders, individually have endeavored to improve healthcare and social care, the complex challenges demands novel and integrative approach to effectively manage the rare diseases scenario (Figure). Additionally, leveraging on technology to create unified platforms and eHealth/mHealth programs can aid quick and better reach. Recommendation: Literature review has established some challenges and approaches to overcome them. However, in order to develop and implement an integrative approach in practice, the current drivers and barriers to effective management must be clearly explored. This demands a sequential explanatory research (quantitative study followed by qualitative) to explain quantitative results on current practices by exploring certain outcomes (especially unexpected ones) in depth. Eventually, an integrated approach can be developed to successfully address concerns and strengthen the management
Shmuel Prints
Clalit Health Service
Israel
Title: The diagnostic challenge of rare diseases: lack of knowledge or leaking method?

Biography:
Shmuel Prints is a certified Internal Diseases specialist with a long-term experience in outpatient practice. He has special interest in rheumatology & lung diseases areas. Dr. Prints acquired his knowledge of statistic methods in medicine from Public Health training in Ben-Gurion University, Israel. This led to his collaboration with prof. Leonid Naumov, a pioneer in algorithm using for medical diagnose. Furthermore, Dr. Prints develops a unique crowd sourcing system for diagnosing rare disorders, based on an understanding of mathematical methods for machine learning. The system's innovation lies in ranking the doctor's presentation of the patient, and not the diagnostic assumptions.
Abstract:
Rare diseases usually take an inordinately long time to diagnose (1,2) This delay is traditionally explained by the low suspicion rate of physicians towards rare disorders, and their lack of knowledge of these items (3,4)
I would like to propose that this does not consider the main culprit of clinical diagnostic workup, algorithmic search.
Methodology & Theoretical Orientation:
The classification algorithm is a step-by-step process for exclusion from all possible entities except right. In every step, it considers one feature of the case. Depending on whether this case displays the feature or not, the algorithm dictates the next explored sign. There are multiple algorithms or “classification trees” for the same signs.
The recognition of diagnostic symptoms and signs in current medicine is never exact. Therefore, any classification tree has missed diagnoses.
For ancient medical traditions and newest insurance imperatives, the best algorithm must have minimal mistakes. The result of this approach is a quick and accurate diagnosis of the most frequent and well-known diseases, and neglect of rare clinical entities.
New approaches to medical diagnosis base on machine learning. They traditionally focus on maximizing the recognition rates and suffer from the same defect (5-7)
Conclusion: To better recognize rare diseases, we need to take a different way to their diagnosis in general.
Alice Abdel Aleem
Weill Cornell Medical College
Qatar
Title: Hereditary Spastic Paraplegias phenotype Constitute Part of Broader Rare Genetic Mendelian Inherited Disorders

Biography:
Dr. Alice Abdel Aleem has her expertise in the field of human clinical and molecular genetics with particular interest in neurogenetics disorders. Her primary area of interest is to provide reliable and high quality research results to health care physicians to improve diagnostics in human genetic disorders. Her current Extramural funded research is focusing on genes identification in monogenetic disorders. She is mainly concerned with building clinical and genomic databases for patients, encountered in Qatar, with spastic paraplegias, heritable muscle diseases, brain malformation, and interesting unrecognized Mendelian disorders. Results of her research is functionally investigated in her lab and in collaboration with investigators in international academic institutes in order to be able to provide confirmed information to health care physicians to use in counseling and managing their patients.
Abstract:
Hereditary Spastic Paraplegias (HSPs) are a group of rare neurological diseases of remarkable clinical and genetic heterogeneity. Cardinal features involve lower limbs spasticity, abnormal gait and difficult walking that eventually ends, in most of the cases, in being a wheel chair bound. Interestingly, presentation in patients with HSPs, particularly the autosomal recessive forms, is much more than lower limbs spasticity and difficult walking. The variable association with developmental delay, psychomotor retardation, learning disabilities or even mental retardation, retinopathy, skin changes, distinctive brain malformation, ataxia, or extrapyramidal involvements brings up AR-HSPs as rare syndromes of broad clinical spectrum rather than just neurodegenerative spastic movement disorders. The axonal transport machinery, altered in HSP, comprises elaborate components of motor proteins, microtubules, shaping and distribution of subcellular organelles and enzymes involved in nucleotides or lipid metabolism.
Families of different ethnic background; Qatari and other ethnicities, with a unified clinical feature of variably progressive lower limbs spasticity and walking difficulty were ascertained. Families either with only these standard features or in association with variable presentations of ataxia, pain insensitivity, remarkable vertebral destruction, regression in mental abilities, severe psychomotor retardation, and notable neuroradiological abnormalities were enrolled in the study. Whole Genome Sequencing (WGS) was applied to identify candidate genes in the recruited families.
Clinical findings presented, in addition to demographic and age groups’ distribution, complex HSP rare phenotypes with interesting extraneural presentations. Of which, features of marked pain insensitivity, skin changes and cerebellar atrophy were seen in independent families. WGS revealed involvements of rarely encountered HSPs genes, of which genes deriving purines and fatty acid metabolism and mitochondrial proteins. A Family with double pathogenic mutations in Parkin gene and a known HSP gene was identified.
Identification of causative genes of rare Mendelian diseases in a research sitting promotes the opportunity to diagnostics molecular genetics, improved genetic counseling qualities and primary prevention.
Marc Dooms
Production manager
Center for Clinical Pharmacology
Belgium
Title: From Promising Molecules to Orphan Drugs: Early Clinical Drug Development

Biography:
Marc Dooms is a Production manager, Center for Clinical Pharmacology, Leuven, Belgium. He is also worked as a Senior Pharmacist at the University Hospitals in Leuven.
Abstract:
Phase-1 (also known as “First-in-Man”) clinical trials initiate the early clinical development of possible new medicines. By raising the dose of the investigational compound in healthy volunteers pharmacokinetic and –dynamic parameters are recorded alongside the safety profile of the new substance in humans. Patient participation in this early phase of clinical trials will be rather limited. After successful phase -1 trials, further phase -2 and phase -3 clinical trials in patients may lead to a marketing authorization.
In the first 15 years of the European Union Orphan Drug Directive 4-5- percent of the orphan drug applications were authorizated. However, for many of these orphan drugs no phase -1 studies were required as these products were already well known pharmaceutical substances with a clearly defined pharmacological profile. Furthermore, for 19 orphan drugs, already authorized by the European Medicines Agency, the original rare indication was extended to another rare disease and no phase -1 trials were needed. For all the other orphan drugs clinical development started with regular phase -1 studies in human volunteers
William S Baek
Parkside Medical Group
USA
Title: Four Rare Neurogenetic Disorders: Underlying Mechanisms and Management

Biography:
Dr. William S Baek is a triple board-certified neurologist. Born in NYC, he graduated from Seoul National University College of Medicine in 1999 and completed his Neurology residency at the University of Chicago and a fellowship in Clinical Neurophysiology at UC San Diego in 2006. He completed an NIH postdoctorate research fellowship at the Children’s Hospital of Philadelphia.
He has served on UM at Beaver Medical Group, and as the Primary Stroke Center Medical Director, Internal Medical Residency Program Faculty Member, Neurology Clerkship Director, EMR champion, and QI/Peer Review Committee member at Kaiser Fontana Medical Center. He was a member of the Donald M. Palatucci Advocacy Leadership Forum, Class 2014. He also serves on the Clerkship Directors Consortium, Ethics Section of the AAN and is a member of the AANEM.
He was the official bilingual moderator for the 2009 AOCCN, IFCN, in Seoul, Korea. He is on the Editorial Board for the Journal of Neurology and Neuroscience and JSM Alzheimer's Disease and Related Dementia. He has over 30 publications, almost all as sole author.
He is a certified medical interpreter for Korean and Spanish he has done TV shows in Korean, English and Spanish. He studied German at Harvard University. He also a professional medical translator for Japanese.
Abstract:
Since the beginning of the 21st century the field of Neurogenetics has exploded, generating novel concepts, unveiling mechanisms, and creating the basis for innovative molecule-targeted specific therapies for neurological disorders. Establishing a genetic diagnosis for any neurological condition is critical for understanding the natural course of the disease and managing accordingly; it shall no longer be viewed as medically unnecessary. This has created a paradigm shift towards reclassifying diseases based on the molecular features rather than signs and symptoms.
Down syndrome, 22q11.2 deletion syndrome, Angelman syndrome, Prader Willi syndrome, Klinefelter syndrome, Turner syndrome, cri-du-chat (5p deletion), phenylketonuria, neurocutaneous disorders, Duchenne’s muscular dystrophy, Friedreich’s ataxia (1/50,000), myotonic dystrophy, Huntington’s disease (1/10,000), and Charcot-Marie-Tooth disease(1/3000) are among the most common hereditary neurological disorders which are fairly well-known.
I would like to present four genetically confirmed cases that are much rarer, with their underlying mechanisms and management in the everyday clinical setting.
These are cases which I have personally diagnosed and treated, such as horizontal gaze palsy with progressive sclerosis(HGPPS), Smith-Magenis syndrome(SMS), X-linked ichthyosis(XLI) and Phelan McDermid (PMD) syndrome, with review of the literature
Yvonne Brenda Nabunnya
Makerere University College of Health Sciences
Uganda
Title: The Safety and Efficacy of Prednisolone In Preventing Re-Accumulation Of Ascites Among Endomyocardial Fibrosis Patients In Uganda: A Randomised Clinical Trial

Biography:
Yvonne Brenda Nabunnya working as a Department of Medicine, Makerere University College of Health Sciences in Uganda
Abstract:
Background
Endomyocardial fibrosis (EMF), the commonest restrictive cardiomyopathy worldwide, is characterized by obliterative inflammation and fibrosis of the endocardium. Inflammation in other parts of the body such as the peritoneum may explain the accumulation of ascites, a painful and disabling feature of this disease. Therefore, we aimed to determine the efficacy and safety of prednisolone to prevent re-accumulation of ascites from International Ascites Club grade 2 to grade 3 among EMF patients attending Mulago hospital cardiology service.
Methods
This was a randomised placebo controlled trial with a 1:1 parallel design. Over a period of ten months, participants were recruited and randomized to receive 1mg/Kg per day of prednisolone or placebo and were followed for a maximum of 8 weeks. The primary outcome was re-accumulation of grade 3 ascites. Safety was assessed by self-reported side effects, physical exam, and laboratory assessment.
Results
Sixteen patients were randomised to prednisolone, while nineteen were randomised to placebo. Six patients were lost to follow up (1-prednisolone arm, 5-placebo). Baseline characteristics were balanced between groups, although only 4% had exudative ascites and only 10% had eosinophilia overall. Prednisolone was safely administered in this setting; however, there was no statistically significant difference in the overall risk of developing grade 3 ascites over 8 weeks (RR (95% confidence interval) 0.70 (0.439-1.114), p=0.12). The rate of the primary outcome per 1000 person days of follow-up was also similar in both arms (p=0.63).
Conclusion
Short term prednisolone use was generally safe in this patient population but there was no statistically significant evidence of efficacy. Additional studies are needed to assess the efficacy of anti-inflammatory treatments to slow progression of this disease.
Nuri M. Shembesh
Department of pediatric and pediatric neurology
Benghazi children Hospital
Title: Prevalence and outcome of Treatable lysosomal storage diseases in Children from north- eastern part of Libya

Biography:
Nuri M. Shembesh is working as a faculty in Department of pediatric and pediatric neurology, Benghazi children Hospital, Benghazi, Libya
Abstract:
Background: Lysosomal storage diseases are important inherited metabolic disorders with major healthcare concern. LSD occur at all ages and are clinically diverse, they differ greatly in their rate of progression and represent a large burden of illness in the population.
The range of manifestations includes organomegaly, disturbed function of visceral organs, skeletal effects and neurological features.
There is no specific or curative treatment for most lysosomal storage diseases, supportive and palliative treatment are nonetheless of great benefit .however recently recombinant DNA technology has led to the development of enzyme –replacement therapy for several lysosomal diseases .
In this paper we present our experience from north-eastern part of Libya with some of this treatable LSD.
Methods: Twenty Seven cases with treatable LSD disorders were diagnosed and followed up in Benghazi children hospital during the period from (1997 – 2013) with Four different treatable LSD and general prevalence rate of 2.7/100.000.
Initial work-up focused on clinical, laboratory and radiological evaluation. Lysosomal enzyme assay in peripheral blood leukocytes were performed according to standard techniques.
Results: Twelve children were diagnosed as Gaucher disease with prevalence rate of 1.2/100.000 The median age at diagnosis was 1 year, male to female ratio 1:1 , Six cases of Gaucher type one four of them on enzyme replacement two brother and sister with mild form on regular follow up without treatment . Four cases of type three all receiving cerozyme as replacement therapy and one child diagnosed as Gaucher type two died after one year of diagnosis he was also in Enzyme replacement therapy none of our patients has bone marrow transplant.
Nine children were diagnosed as MPS1 with prevalence rate of 0.9/100.000 the median age at diagnosis was three years, male to female ratio 1.2:1, four have severe MPS1, three were moderate to severe form and one was mild form all of them received alldrozyme replacement therapy non had bone marrow transplant, of severe form one died and one lost follow up.
Three brothers ,two of them were twin Diagnosed as MPS2 (Hunter) with low prevalence rate 0.3/100.000 all from Tobrok in the far east point of Libya ,non of them received enzyme replacement therapy till now .
Three children were diagnosed as Pompe disease, with prevalence rate of 0.3/100,000.
One patient was suspected as infantile pompe at one and half month of age died before result of enzyme assay. Two brothers diagnosed as juvenile pompe not on enzyme replacement therapy yet.
Conclusion: Lysosomal storage diseases, especially Gaucher and MPS1 disease, may represent important pathologies in our population and their prevalence rate was similar to the reported prevalence rate from other parts of the world .Specific diagnosis and follow-up is the key step in the accurate management and treatment of these patients