Day 2 :
Keynote Forum
Stefano Giacomini
Rizzoli Orthopedic Institute
Italy
Keynote: Neurofibromatosis and Spinal Deformities: The severity in pediatric age and treatment

Biography:
Dr. Stefano Giacomini is a Medical Director; performs orthopedic examinations in Free Job regime at the Outpatient Clinic of Rizzoli. He has done his Specialization in Orthopedics and Traumatology achieved in 2001 at the University of Bologna. He is the author of over 50 scientific publications. He has participated, even as a speaker at dozens of national and international conferences and 'socio GIS (Italian Scoliosis Group)
Abstract:
Background
Scoliosis in neurofibromatosis type I (NF-I) ranges from 10% to 60%; can be divided into three groups 1) dystrophic scoliosis, 2) non-dystrophic scoliosis and 3) early onset scoliosis. Surgical treatment is very demanding in early onset, severely progressive spinal column deformities.
Materials and methods
Twenty-three patients, aged between 4 and 11 years, were surgically treated at the Authors’ Spine Surgery Division in the past 15 years. Mean follow-up is 5 years (range, 18 months to 15 years). Mean age at the time of surgical procedure was 9.1 years (range, 4 years to 11 years). Average scoliosis was 48° (range, 38° to 82°) and skeletal maturity according to Risser sign was 0 in all of the patients. Patients were divided into 2 Groups according to the surgical procedure adopted. Posterior only instrumentation was performed in 16 patients that presented with a thoracic kyphosis lower than 50° (Group A), in the remaining 7 patients showing thoracic kyphosis exceeding 50°, combined anterior and posterior instrumented arthrodesis was performed (Group B). One patient, belonging to Group A, was instrumented with growing rod without fusion.
Results
Average correction of scoliosis was 60%, overall complication rate 24% and major 7%. Crankshaft phenomenon was observed in 21% (Group A): in these cases, anterior arthrodesis was performed after a mean 15-month from first surgical procedure. Fusion failure was observed in 1 (Group B) patient who underwent revision of posterior instrumentation. Clinical and radiographic evaluation at F-up showed good outcome in terms of deformity progression and quality of life.
Conclusion
Early and aggressive surgery is the most effective management for dystrophic curves in neurofibromatosis has been proven to be. Our experience confirms the need for spinal stabilization even in pediatric age in rapidly progressive spinal deformities. The growing rod technique should be carefully evaluated in each single case.
- Rare Hereditary Diseases| Rare Ophthalmological Diseases | Genetic Diseases and Disorders |Strategies for Diagnosis &Treatment| Undiagnosed Rare Diseases
Location: London, UK

Chair
Patrick J Tighe
University of Nottingham
UK
Session Introduction
Jong Wook Chang
Samsung Medical Center
South Korea
Title: Paracrine Action of Human Mesenchymal Stem Cells for Muscle diseases.

Biography:
Jong Wook Chang has his expertise in translational and clinical research of stem cells for neurological diseases including CNS and PNS. Especially, he has made effort to identify soluble factors secreted from human mesenchymal stem cells (MSC) to understand therapeutic effect of MSC. In addition, now he is responsible for management of cGMP facility in Samsung Medical Center to produce clinical grade of MSC for clinical trials.
Abstract:
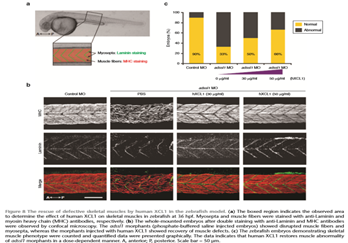
The role of Wharton’s jelly-derived human mesenchymal stem cells (WJ-MSCs) in inhibiting muscle cell death has been elucidated in the present study. Apoptosis induced by serum-deprivation in mouse myoblast cell lines (C2C12) was significantly reduced when the cell lines where co-cultured with WJ-MSCs in a transwell system. Antibody arrays indicated high levels of chemokine (C motif) ligand (XCL1) secretion by co-cultured WJ-MSCs and XCL1 protein treatment resulted in complete inhibition of apoptosis in serum-starved C2C12 cells. Apoptosis of C2C12 cells and loss of differentiated C2C12 myotubes induced by lovastatin, another muscle cell death inducer, was also inhibited by XCL1 treatment. However, XCL1 treatment did not inhibit apoptosis of cell lines other than C2C12. When XCL1-siRNA pretreated WJ-MSCs were co-cultured with serum-starved C2C12 cells, apoptosis was not inhibited, thus confirming that XCL1 is a key factor in preventing C2C12 cell apoptosis. We demonstrated the therapeutic effect of XCL1 on the zebrafish myopathy model, generated by knock down of a causative gene ADSSL1 encoding a muscle isozyme of adenylosuccinate synthase. The exogenous expression of XCL1 resulted in significant recovery of the zebrafish skeletal muscle defects. These results suggest that human WJ-MSCs and XCL1 protein may act as pro-mixing and novel therapeutic agents for treatment of myopathies and other skeletal muscle diseases.
Rob W.J. Collin
Dept. of Human Genetics
Radboud University Medical Center
Title: Splice modulation therapy for inherited retinal diseases

Biography:
Rob W.J. Collin is an Associate Professor at the Department of Human Genetics, at the Radboud University Medical Center in Nijmegen, The Netherlands, and is affiliated to the Donders Institute for Brain, Cognition and Behaviour. During his post-doc, he was involved in the identification of several novel genes underlying inherited hearing impairment, and inherited retinal diseases. Since 2010, he switched his research focus towards the development of Molecular Therapies for Inherited Eye Disorders, in particular those that involve the modulation of pre-mRNA splicing. In 2010, he worked in the lab of Prof. Dr. Jean Bennett, a pioneer in the development of retinal gene therapy. Rob Collin was awarded with a prestigious VENI Award from the Dutch Organization of Scientific Research (2010), an Individual Investigator Award from the Foundation Fighting Blindness (2012), and recently received the IJMS Young Investigator Award (2017).
Abstract:
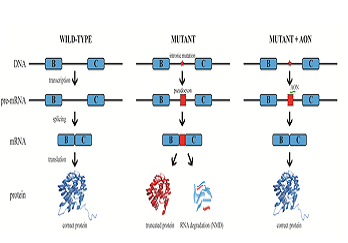
Statement of the Problem: Inherited retinal diseases (IRDs) are characterized by severe and progressive visual impairment, often leading to complete blindness. IRDs display a tremendous genetic and clinical heterogeneity, and to date, no effective treatment exists. A significant fraction of the genetic defects underlying IRD affect pre-mRNA splicing of the corresponding gene. We therefore decided to employ antisense oligonucleotides (AONs) to develop splice modulation therapy for specific genetic subtypes of IRD. AONs are small and versatile DNA/RNA molecules that bind complementary to their target pre-mRNA molecule and are able to redirect pre-mRNA splicing.
Methodology & Theoretical Orientation: Fibroblast cell lines derived from patients harboring mutations that affect pre-mRNA splicing were generated and cultured in the presence of AONs that are specifically designed to correct pre-mRNA splicing. In some cases, fibroblasts were first reprogrammed to induced pluripotent stem cells that were subsequently differentiated to photoreceptor precursor cells, and then treated with AONs. RT-PCR analysis was performed to study the efficacy of AON treatment, and for some cell lines, Western blot analysis or immunocytochemical analysis was performed to determine the effects at the protein and cellular level.
Findings: AON administration to patient-derived fibroblast cells harboring a recurrent splice mutation underlying severe early-onset IRD, i.e. c.2991+1655A>G in CEP290, resulted in full restoration of CEP290 pre-mRNA splicing, a significant increase in CEP290 protein levels, and rescue of a ciliary defect. In addition, AON delivery to patient cells with splice mutations in ABCA4 resulted in correction of the splice defects. Studies targeting additional mutations affecting pre-mRNA splicing that underlie IRD are currently ongoing.
Conclusion & Significance: Splice modulation therapies represent a promising and attractive strategy for the future treatment of specific genetic subtypes of IRD.
Gayathri Balasubramanian
Focus Scientific Research Center (FSRC)
phamax
India
Title: Management of rare diseases: An integrated approach to break down barriers and facilitate patient access to healthcare

Biography:
Dr. Gayathri Balasubramanian is a Scientific Advisor at FSRC, part of phamax AG. She holds a Masters in Clinical Research from the University of Sheffield, UK. She was awarded the “Graduate Award” and “Skills for Work” by the University of Sheffield and other top global organizations for academic, volunteering and extracurricular activities during her tenure there. She has 4 years of progressive experience in the healthcare research domain and she is proficient in primary and secondary research (various assignments employing mixed methods research), systematic reviews and critical appraisal, stakeholders identification, liaison and engagement and publications. She has worked on diverse projects in market assessment, market access, policy shaping, forecasting and epidemiology in therapeutic areas of rare diseases, oncology, diabetes, cardiovascular diseases and infectious diseases.
Email: gayathri.balasubramanian@fs-researchcenter.com
Abstract:
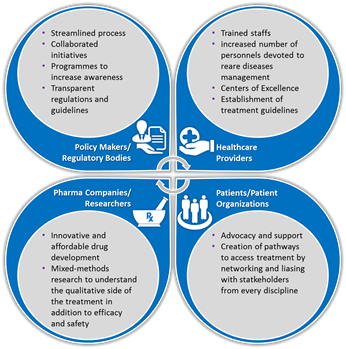
Background: In Europe, a rare disease is a disease that affects less than 1 in 2000. Although this seems to affect a fraction of the population, collectively it imposes a colossal. Rare diseases are disabling, dramatically reducing the quality of life, hampering routine activities and autonomous survival. Rare diseases management is intricate and varies across geographies. Understanding the complexities is important to resolve issues and improve care. Methodology: Secondary research of published and grey literature. Findings: Some of the challenges encountered in rare diseases management are delayed or misdiagnosis, lack of awareness on diseases/management, lack of quality healthcare at proximity, struggles to continued care, treatment access, and social consequences. In recent times, stakeholders such us healthcare providers, patient organizations, pharmaceutical companies, regulatory bodies and policy makers have taken many initiatives to resolve these challenges. National polices/plans are devised exclusively to manage rare diseases. Compassionate use programmes have encouraged research and provided accelerated entry of orphan drugs into the market. Organizations like EURORDIS have given voice to more than 300 rare disease organizations in over 30 countries. While these stakeholders, individually have endeavored to improve healthcare and social care, the complex challenges demands novel and integrative approach to effectively manage the rare diseases scenario (Figure). Additionally, leveraging on technology to create unified platforms and eHealth/mHealth programs can aid quick and better reach. Recommendation: Literature review has established some challenges and approaches to overcome them. However, in order to develop and implement an integrative approach in practice, the current drivers and barriers to effective management must be clearly explored. This demands a sequential explanatory research (quantitative study followed by qualitative) to explain quantitative results on current practices by exploring certain outcomes (especially unexpected ones) in depth. Eventually, an integrated approach can be developed to successfully address concerns and strengthen the management
Shmuel Prints
Clalit Health Service
Israel
Title: The diagnostic challenge of rare diseases: lack of knowledge or leaking method?

Biography:
Shmuel Prints is a certified Internal Diseases specialist with a long-term experience in outpatient practice. He has special interest in rheumatology & lung diseases areas. Dr. Prints acquired his knowledge of statistic methods in medicine from Public Health training in Ben-Gurion University, Israel. This led to his collaboration with prof. Leonid Naumov, a pioneer in algorithm using for medical diagnose. Furthermore, Dr. Prints develops a unique crowd sourcing system for diagnosing rare disorders, based on an understanding of mathematical methods for machine learning. The system's innovation lies in ranking the doctor's presentation of the patient, and not the diagnostic assumptions.
Abstract:
Rare diseases usually take an inordinately long time to diagnose (1,2) This delay is traditionally explained by the low suspicion rate of physicians towards rare disorders, and their lack of knowledge of these items (3,4)
I would like to propose that this does not consider the main culprit of clinical diagnostic workup, algorithmic search.
Methodology & Theoretical Orientation:
The classification algorithm is a step-by-step process for exclusion from all possible entities except right. In every step, it considers one feature of the case. Depending on whether this case displays the feature or not, the algorithm dictates the next explored sign. There are multiple algorithms or “classification trees” for the same signs.
The recognition of diagnostic symptoms and signs in current medicine is never exact. Therefore, any classification tree has missed diagnoses.
For ancient medical traditions and newest insurance imperatives, the best algorithm must have minimal mistakes. The result of this approach is a quick and accurate diagnosis of the most frequent and well-known diseases, and neglect of rare clinical entities.
New approaches to medical diagnosis base on machine learning. They traditionally focus on maximizing the recognition rates and suffer from the same defect (5-7)
Conclusion: To better recognize rare diseases, we need to take a different way to their diagnosis in general.
Alice Abdel Aleem
Weill Cornell Medical College
Qatar
Title: Hereditary Spastic Paraplegias phenotype Constitute Part of Broader Rare Genetic Mendelian Inherited Disorders

Biography:
Dr. Alice Abdel Aleem has her expertise in the field of human clinical and molecular genetics with particular interest in neurogenetics disorders. Her primary area of interest is to provide reliable and high quality research results to health care physicians to improve diagnostics in human genetic disorders. Her current Extramural funded research is focusing on genes identification in monogenetic disorders. She is mainly concerned with building clinical and genomic databases for patients, encountered in Qatar, with spastic paraplegias, heritable muscle diseases, brain malformation, and interesting unrecognized Mendelian disorders. Results of her research is functionally investigated in her lab and in collaboration with investigators in international academic institutes in order to be able to provide confirmed information to health care physicians to use in counseling and managing their patients.
Abstract:
Hereditary Spastic Paraplegias (HSPs) are a group of rare neurological diseases of remarkable clinical and genetic heterogeneity. Cardinal features involve lower limbs spasticity, abnormal gait and difficult walking that eventually ends, in most of the cases, in being a wheel chair bound. Interestingly, presentation in patients with HSPs, particularly the autosomal recessive forms, is much more than lower limbs spasticity and difficult walking. The variable association with developmental delay, psychomotor retardation, learning disabilities or even mental retardation, retinopathy, skin changes, distinctive brain malformation, ataxia, or extrapyramidal involvements brings up AR-HSPs as rare syndromes of broad clinical spectrum rather than just neurodegenerative spastic movement disorders. The axonal transport machinery, altered in HSP, comprises elaborate components of motor proteins, microtubules, shaping and distribution of subcellular organelles and enzymes involved in nucleotides or lipid metabolism.
Families of different ethnic background; Qatari and other ethnicities, with a unified clinical feature of variably progressive lower limbs spasticity and walking difficulty were ascertained. Families either with only these standard features or in association with variable presentations of ataxia, pain insensitivity, remarkable vertebral destruction, regression in mental abilities, severe psychomotor retardation, and notable neuroradiological abnormalities were enrolled in the study. Whole Genome Sequencing (WGS) was applied to identify candidate genes in the recruited families.
Clinical findings presented, in addition to demographic and age groups’ distribution, complex HSP rare phenotypes with interesting extraneural presentations. Of which, features of marked pain insensitivity, skin changes and cerebellar atrophy were seen in independent families. WGS revealed involvements of rarely encountered HSPs genes, of which genes deriving purines and fatty acid metabolism and mitochondrial proteins. A Family with double pathogenic mutations in Parkin gene and a known HSP gene was identified.
Identification of causative genes of rare Mendelian diseases in a research sitting promotes the opportunity to diagnostics molecular genetics, improved genetic counseling qualities and primary prevention.
Marc Dooms
Production manager
Center for Clinical Pharmacology
Belgium
Title: From Promising Molecules to Orphan Drugs: Early Clinical Drug Development

Biography:
Marc Dooms is a Production manager, Center for Clinical Pharmacology, Leuven, Belgium. He is also worked as a Senior Pharmacist at the University Hospitals in Leuven.
Abstract:
Phase-1 (also known as “First-in-Man”) clinical trials initiate the early clinical development of possible new medicines. By raising the dose of the investigational compound in healthy volunteers pharmacokinetic and –dynamic parameters are recorded alongside the safety profile of the new substance in humans. Patient participation in this early phase of clinical trials will be rather limited. After successful phase -1 trials, further phase -2 and phase -3 clinical trials in patients may lead to a marketing authorization.
In the first 15 years of the European Union Orphan Drug Directive 4-5- percent of the orphan drug applications were authorizated. However, for many of these orphan drugs no phase -1 studies were required as these products were already well known pharmaceutical substances with a clearly defined pharmacological profile. Furthermore, for 19 orphan drugs, already authorized by the European Medicines Agency, the original rare indication was extended to another rare disease and no phase -1 trials were needed. For all the other orphan drugs clinical development started with regular phase -1 studies in human volunteers
William S Baek
Parkside Medical Group
USA
Title: Four Rare Neurogenetic Disorders: Underlying Mechanisms and Management

Biography:
Dr. William S Baek is a triple board-certified neurologist. Born in NYC, he graduated from Seoul National University College of Medicine in 1999 and completed his Neurology residency at the University of Chicago and a fellowship in Clinical Neurophysiology at UC San Diego in 2006. He completed an NIH postdoctorate research fellowship at the Children’s Hospital of Philadelphia.
He has served on UM at Beaver Medical Group, and as the Primary Stroke Center Medical Director, Internal Medical Residency Program Faculty Member, Neurology Clerkship Director, EMR champion, and QI/Peer Review Committee member at Kaiser Fontana Medical Center. He was a member of the Donald M. Palatucci Advocacy Leadership Forum, Class 2014. He also serves on the Clerkship Directors Consortium, Ethics Section of the AAN and is a member of the AANEM.
He was the official bilingual moderator for the 2009 AOCCN, IFCN, in Seoul, Korea. He is on the Editorial Board for the Journal of Neurology and Neuroscience and JSM Alzheimer's Disease and Related Dementia. He has over 30 publications, almost all as sole author.
He is a certified medical interpreter for Korean and Spanish he has done TV shows in Korean, English and Spanish. He studied German at Harvard University. He also a professional medical translator for Japanese.
Abstract:
Since the beginning of the 21st century the field of Neurogenetics has exploded, generating novel concepts, unveiling mechanisms, and creating the basis for innovative molecule-targeted specific therapies for neurological disorders. Establishing a genetic diagnosis for any neurological condition is critical for understanding the natural course of the disease and managing accordingly; it shall no longer be viewed as medically unnecessary. This has created a paradigm shift towards reclassifying diseases based on the molecular features rather than signs and symptoms.
Down syndrome, 22q11.2 deletion syndrome, Angelman syndrome, Prader Willi syndrome, Klinefelter syndrome, Turner syndrome, cri-du-chat (5p deletion), phenylketonuria, neurocutaneous disorders, Duchenne’s muscular dystrophy, Friedreich’s ataxia (1/50,000), myotonic dystrophy, Huntington’s disease (1/10,000), and Charcot-Marie-Tooth disease(1/3000) are among the most common hereditary neurological disorders which are fairly well-known.
I would like to present four genetically confirmed cases that are much rarer, with their underlying mechanisms and management in the everyday clinical setting.
These are cases which I have personally diagnosed and treated, such as horizontal gaze palsy with progressive sclerosis(HGPPS), Smith-Magenis syndrome(SMS), X-linked ichthyosis(XLI) and Phelan McDermid (PMD) syndrome, with review of the literature
Yvonne Brenda Nabunnya
Makerere University College of Health Sciences
Uganda
Title: The Safety and Efficacy of Prednisolone In Preventing Re-Accumulation Of Ascites Among Endomyocardial Fibrosis Patients In Uganda: A Randomised Clinical Trial

Biography:
Yvonne Brenda Nabunnya working as a Department of Medicine, Makerere University College of Health Sciences in Uganda
Abstract:
Background
Endomyocardial fibrosis (EMF), the commonest restrictive cardiomyopathy worldwide, is characterized by obliterative inflammation and fibrosis of the endocardium. Inflammation in other parts of the body such as the peritoneum may explain the accumulation of ascites, a painful and disabling feature of this disease. Therefore, we aimed to determine the efficacy and safety of prednisolone to prevent re-accumulation of ascites from International Ascites Club grade 2 to grade 3 among EMF patients attending Mulago hospital cardiology service.
Methods
This was a randomised placebo controlled trial with a 1:1 parallel design. Over a period of ten months, participants were recruited and randomized to receive 1mg/Kg per day of prednisolone or placebo and were followed for a maximum of 8 weeks. The primary outcome was re-accumulation of grade 3 ascites. Safety was assessed by self-reported side effects, physical exam, and laboratory assessment.
Results
Sixteen patients were randomised to prednisolone, while nineteen were randomised to placebo. Six patients were lost to follow up (1-prednisolone arm, 5-placebo). Baseline characteristics were balanced between groups, although only 4% had exudative ascites and only 10% had eosinophilia overall. Prednisolone was safely administered in this setting; however, there was no statistically significant difference in the overall risk of developing grade 3 ascites over 8 weeks (RR (95% confidence interval) 0.70 (0.439-1.114), p=0.12). The rate of the primary outcome per 1000 person days of follow-up was also similar in both arms (p=0.63).
Conclusion
Short term prednisolone use was generally safe in this patient population but there was no statistically significant evidence of efficacy. Additional studies are needed to assess the efficacy of anti-inflammatory treatments to slow progression of this disease.
Nuri M. Shembesh
Department of pediatric and pediatric neurology
Benghazi children Hospital
Title: Prevalence and outcome of Treatable lysosomal storage diseases in Children from north- eastern part of Libya

Biography:
Nuri M. Shembesh is working as a faculty in Department of pediatric and pediatric neurology, Benghazi children Hospital, Benghazi, Libya
Abstract:
Background: Lysosomal storage diseases are important inherited metabolic disorders with major healthcare concern. LSD occur at all ages and are clinically diverse, they differ greatly in their rate of progression and represent a large burden of illness in the population.
The range of manifestations includes organomegaly, disturbed function of visceral organs, skeletal effects and neurological features.
There is no specific or curative treatment for most lysosomal storage diseases, supportive and palliative treatment are nonetheless of great benefit .however recently recombinant DNA technology has led to the development of enzyme –replacement therapy for several lysosomal diseases .
In this paper we present our experience from north-eastern part of Libya with some of this treatable LSD.
Methods: Twenty Seven cases with treatable LSD disorders were diagnosed and followed up in Benghazi children hospital during the period from (1997 – 2013) with Four different treatable LSD and general prevalence rate of 2.7/100.000.
Initial work-up focused on clinical, laboratory and radiological evaluation. Lysosomal enzyme assay in peripheral blood leukocytes were performed according to standard techniques.
Results: Twelve children were diagnosed as Gaucher disease with prevalence rate of 1.2/100.000 The median age at diagnosis was 1 year, male to female ratio 1:1 , Six cases of Gaucher type one four of them on enzyme replacement two brother and sister with mild form on regular follow up without treatment . Four cases of type three all receiving cerozyme as replacement therapy and one child diagnosed as Gaucher type two died after one year of diagnosis he was also in Enzyme replacement therapy none of our patients has bone marrow transplant.
Nine children were diagnosed as MPS1 with prevalence rate of 0.9/100.000 the median age at diagnosis was three years, male to female ratio 1.2:1, four have severe MPS1, three were moderate to severe form and one was mild form all of them received alldrozyme replacement therapy non had bone marrow transplant, of severe form one died and one lost follow up.
Three brothers ,two of them were twin Diagnosed as MPS2 (Hunter) with low prevalence rate 0.3/100.000 all from Tobrok in the far east point of Libya ,non of them received enzyme replacement therapy till now .
Three children were diagnosed as Pompe disease, with prevalence rate of 0.3/100,000.
One patient was suspected as infantile pompe at one and half month of age died before result of enzyme assay. Two brothers diagnosed as juvenile pompe not on enzyme replacement therapy yet.
Conclusion: Lysosomal storage diseases, especially Gaucher and MPS1 disease, may represent important pathologies in our population and their prevalence rate was similar to the reported prevalence rate from other parts of the world .Specific diagnosis and follow-up is the key step in the accurate management and treatment of these patients